为重症救治赋能
为患者康复加速

登录
注册
登录方式
方式一:
PC端网页:www.rccrc.cn
输入账号密码登录,可将此网址收藏并保存密码方便下次登录
方式二:
手机端网页:www.rccrc.cn
输入账号密码登录,可将此网址添加至手机桌面并保存密码方便下次登录
方式三:
【重症肺言】微信公众号
输入账号密码登录
注:账号具有唯一性,即同一个账号不能在两个地方同时登录。
孙庆文,梁微波,徐远达,黎毅敏 广州医科大学附属第一医院呼吸与危重症医学科 发布于2023-03-22 浏览 6740
 收藏
收藏
作者:孙庆文,梁微波,徐远达,黎毅敏
临床资料
病史情况
患者女性,17岁,学生,入院8个月前出现双下肢凹陷性水肿,伴乏力,下肢肌肉酸痛,间伴心悸,多次于外院就诊查肌酶升高,肌电图提示“肌源性损害”,拟“多发性肌炎,格林-巴利综合征”,予大剂量激素、丙种球蛋白冲击、羟氯喹、环磷酰胺(CTX)化疗、营养神经等治疗,症状缓解不明显。因上述症状加重1周于2012年12月4日就诊于广州医科大学第一附属医院风湿科。入院初步诊断:多发性肌炎?肺炎。
脉搏125次/min,双手背轻度浮肿,双下肢非凹陷性水肿,双上肢肌力4级,双下肢肌力3级。多次血乳酸>12.0 mmol/L(正常值:0.7~2.1 mmol/L);血气分析示pH 7.235~7.405,HCO3- 8.2~14.7 mmol/L;肌酸激酶(CK)734 U/L(正常值:10~190 U/L),乳酸脱氢酶(LDH)1274 U/L(正常值:109~255 U/L);外院胸部X线片未见异常;心电图示窦性心动过速,T波改变;心脏彩色超声+心功能:三尖瓣反流(中度),肺动脉高压(48 mmHg),左室收缩功能未见异常,心包少量积液,射血分数(EF)60%;外院肌电图示肌源性损害。
患者入院5天后逐渐出现端坐呼吸,气促明显,B型脑钠尿肽前体(pro-BNP)3294 pg/ml(正常值:<155 pg/ml),仍顽固性高乳酸血症,血气分析提示代谢性酸中毒并呼吸性碱中毒,pH 7.32,PaCO2 18.9 mmHg,PaO2 121 mmHg,HCO3- 9.4 mmol/L,床边胸部X线片提示心影增大,两肺渗出,感染合并肺水肿,两侧少量胸腔积液。
入院7 d后转入该院重症医学科。转入时患者四肢乏力,活动后气促,查体:呼吸30次/min,SpO2 100%(3 L/min吸氧下),双肺呼吸音清,双下肺可闻及湿啰音。心率120~138次/min,四肢非凹陷性水肿。神经系统检查:四肢肌肉萎缩明显,近端肌力2级,远端肌力4级,肌张力正常,病理征阴性。WBC较正常值升高,予持续无创通气,加强抗感染治疗,患者乏力症状进行性加重,出现咳痰无力,咀嚼、吞咽、抬头困难,于入院14 d后行气管插管接呼吸机辅助呼吸,出现血压下降,尿少,不除外感染性休克,行床边连续静脉-静脉血液滤过(CVVH),但患者仍有顽固性高乳酸血症表现,深部痰涂片有细菌、真菌菌丝和孢子,机械通气1周后考虑拔管困难,遂行气管切开。
病情稳定阶段:持续CVVH甚至部分高容量血液滤过(HVHF),血乳酸下降仍不明显,除气管插管时镇静后血压略下降外,不需血管活性药维持血压,有创机械通气后患者氧合情况明显好转,胸部X线片吸收快,肺部感染很快得到控制,但全身肌无力明显并撤机困难。自身免疫指标检查结果均为阴性,不支持结缔组织疾病,重症医学科提供了可靠的支持治疗平台,考虑既往有8个月进行性肌无力病史,再次行肌电图、股四头肌肌肉活检,光镜下病理以肌肉萎缩为主,无血管炎表现,炎症细胞少。
拟“代谢性肌病、脂质沉积性肌病未排除”予辅酶Q10 100 mg bid、左旋肉毒碱 2g qd、维生素B2 10 mg tid、丙种球蛋白0.4 g/kg iv drip qd*5天,小剂量皮质激素及调整富含肉毒碱饮食等治疗。
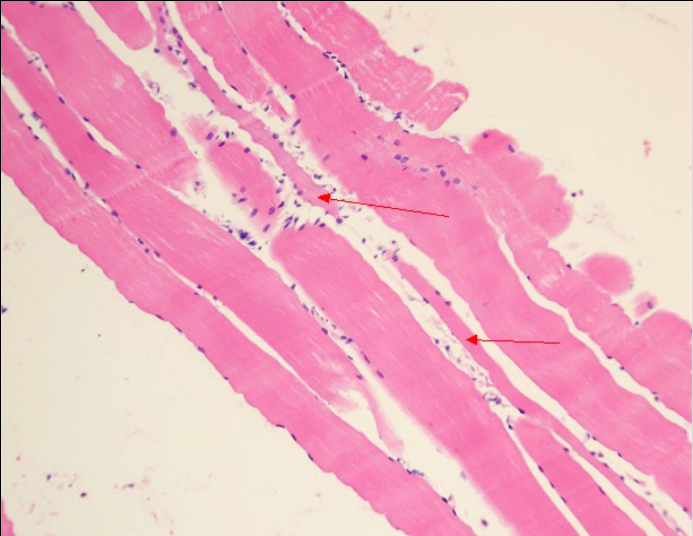
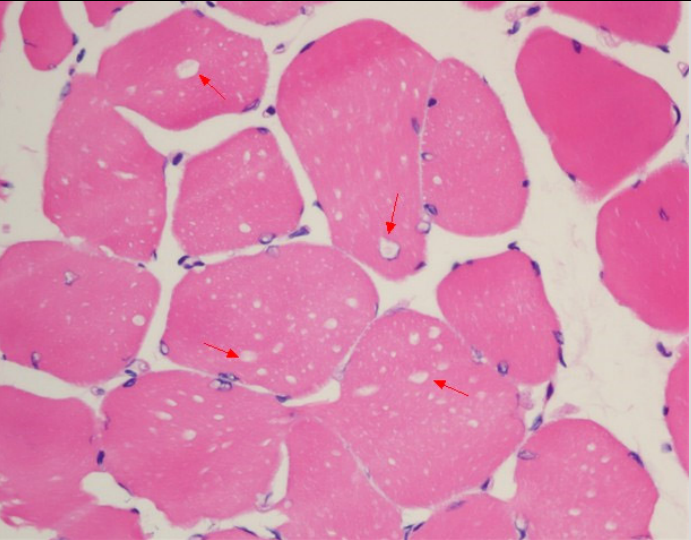
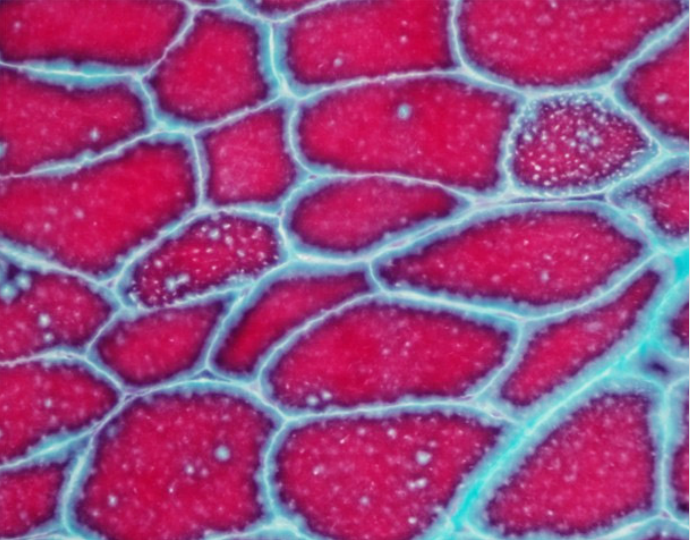
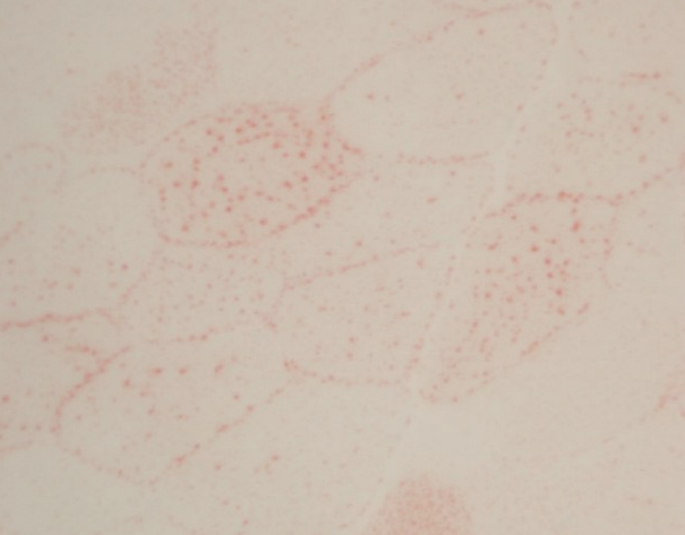
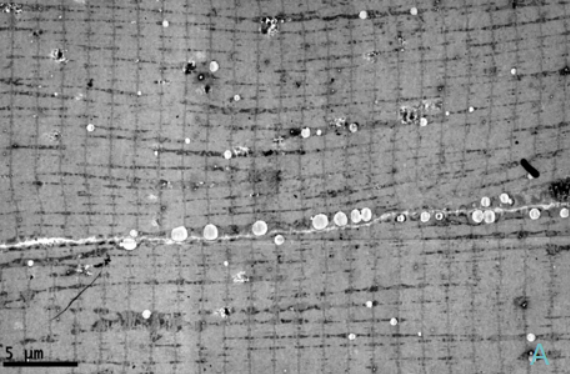
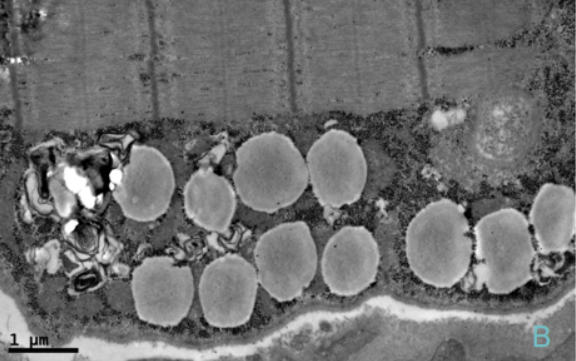
治疗4周后患者四肢近端肌力3级,双手可上举至头,远端肌力5级,间断停用呼吸机。代谢性酸中毒得以纠正,血乳酸水平波动于7~8 mmol/L。复查:CK 277 U/L,LDH 501 U/L。股四头肌肌肉病理见图1~图5。
讨论
参考文献 [1] Wang Z Q, Chen X J, Murong S X, et al. Molecular analysis of 51 unrelated pedigrees with late-onset multiple acyl-CoA dehydrogenation deficiency (MADD)in southern China confirmed the most common ETFDH mutation and high carrier frequency of c.250G>A[J]. J Mol Med (Berl), 2011, 89:569-576. [2] Yan C Z, Lu J H. The pathogenesis research progress of lipid storage myopathy in China[J]. Clin Neuropathol, 2011, 44(5):300-303. [3] Olsen R K, Pourfarzam M, M orris A A, et al. Lipid-storage myopathy and respiratory insufficiency due to ETFQO mutations in a patient with late-onset multiple acyl-CoA dehydrogenation deficiency[J]. J Inherit Metab Dis, 2004, 27(5):671-678. [4] Xi J Y, Lu J H, Zhao C B, et al. The analysis of clinical characteristics and ETFDH gene mutations in 35 lipid storage myopathy cases[J]. Chinese Journal of Neurology, 2011, 44(5):314-321. [5] Rivers E, Nguyen B, Havstad S, et al. Early goal directed therapy in the treatment of severe sepsis and septic shock[J]. N Engl J Med, 2001, 345(19):1368-1377. [6] Jansen T C, van Bommel J, Woodward R, et al. Association between blood lactate levels, Sequential Organ Failure Assessment subscores, and 28-day mortality during early and late intensive care unit stay: a retrospective observational study[J]. Crit Care Med, 2009, 37(8):2369-2374. [7] Gunnerson K J, Saul M, He S, et al. Lactate versus non-lactate metabolic acidosis: a retrospective outcome evaluation of critically ill patients[J]. Crit Care, 2006, 10(1):R22. [8] Gutierrez G, Wulf M E. Lactic acidosis in sepsis: another commentary[J]. Crit Care Med, 2005, 33(10):2420-2422. [9] Wang Y, Zhao D H, Hong D J, et al. There are 20 family genes of ETFDH exist hotspot mutations in Riboflavin responsive lipid storage myopathy patients[J]. Chinese Journal of Neurology, 2011, 44:309-313. [10] Wen B, Dai T, Li W, et al. Riboflavin-responsive lipid-storage myopathy caused by ETFDH gene mutations[J]. J Neurol Neurosurg Psychiatry, 2010, 81:231-236. [11] Li W, Yan C Z, Wu J L, et al. Clinic treatment and Prognosis of follow-up in 42 lipid storage myopathy patients[J]. Chinese Journal of Neurology, 2007, 40(4):229-231. [12] Wu Z Y, Wang N. The status and hot topics of Riboflavin reactive lipid storage myopathy gene research in China[J]. Chinese Journal of Neurology, 2011, 44:297-299. [13] Schoser B G, Pongratz D. Extraocular mitochondrial myopathies and their differential diagnoses[J]. Strabismus, 2006, 14(2):107-113. [10] Gong L Y, Hu F, Huang G, et al. The analysis of clinical manifestations and electro-neurophysiology in lipid storage myopathy patients[J]. Chinese Journal of Neuroimmunology and Neurology, 2013, 20(1):6-8.
【注:本文仅用于学术内容的传播和交流,不用于任何商业和推广】
 后可发表评论
后可发表评论




友情链接
联系我们
 公众号
公众号
 客服微信
客服微信