为重症救治赋能
为患者康复加速

登录
注册
登录方式
方式一:
PC端网页:www.rccrc.cn
输入账号密码登录,可将此网址收藏并保存密码方便下次登录
方式二:
手机端网页:www.rccrc.cn
输入账号密码登录,可将此网址添加至手机桌面并保存密码方便下次登录
方式三:
【重症肺言】微信公众号
输入账号密码登录
注:账号具有唯一性,即同一个账号不能在两个地方同时登录。
周华 王珺 刘超 浙江大学医学院附属第一医院 杰毅生物 发布于2022-02-16 浏览 6239
 收藏
收藏
【摘要】 病原宏基因组测序(mNGS)通过对临床疑似感染患者标本中提取的总核酸进行无偏倚的鸟枪法测序(shotgun sequencing),获得的测序数据先过滤人源序列,再与微生物基因组数据库进行比对分析,汇报样本中的微生物种类和序列数(最佳比对或严格比对至某微生物属/种的核酸片段数)。因为测序过程是无偏倚的,理论上可以覆盖样本中所有已知基因序列的微生物,因此mNGS可以检测上万种病原微生物(细菌、病毒、真菌、寄生虫),具有常规靶向检测(微生物培养、抗体/抗原、PCR、16S/18S测序等)所不具备的无需假设、广覆盖的优点。
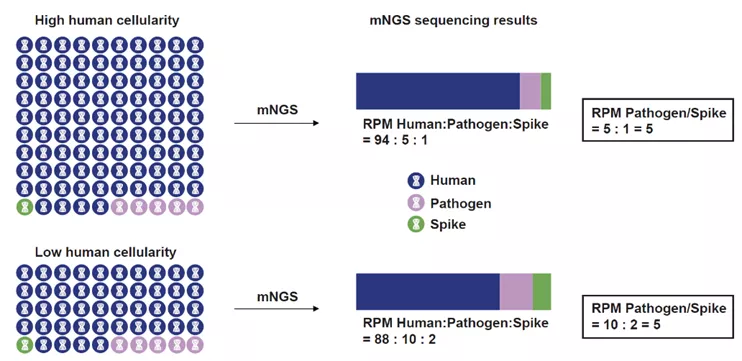
宏基因组测序(mNGS)通常汇报微生物的序列数(序列数reads或每百万reads中的序列数RPM),然而,mNGS的测序过程是对文库中的核酸进行无偏好性的等比例抽样,最终测序的核酸只占文库中所有核酸的千分之一甚至更低[1],因此,mNGS对样本中微生物的检测性能取决于两个因素:人源核酸含量与病原核酸含量。人源核酸含量越高,微生物的检测性能越差。在恒定的测序数据量下(共识推荐20M reads[2]),人源核酸含量高的文库中,相同含量的微生物序列数会低于人源核酸含量低的文库。为解决临床标本中人源核酸对微生物检测的影响,去宿主/去人源技术被广泛用于临床mNGS检测,目前主要的去宿主技术是差异化裂解,通过皂苷(saponin)一类的化学物质选择性裂解人细胞,再通过核酸酶处理释放出的人源核酸,从而对微生物进行富集[3]。但这种处理方式可能会造成某些微生物的损失(比如肺炎链球菌),从而产生假阴性结果[4]。在本研究中,我们通过向肺泡灌洗液中加入内标核酸分子,对每份标本中的人源核酸和微生物核酸进行相对定量,从而对人源含量和病原含量实现标化(Q-mNGS)。我们还对Q-mNGS的检验性能与诊断性能在205例疑似下呼吸道感染的患者中进行了前瞻性的临床研究,同时比较了去宿主前后微生物检测性能的差异。
材料和方法
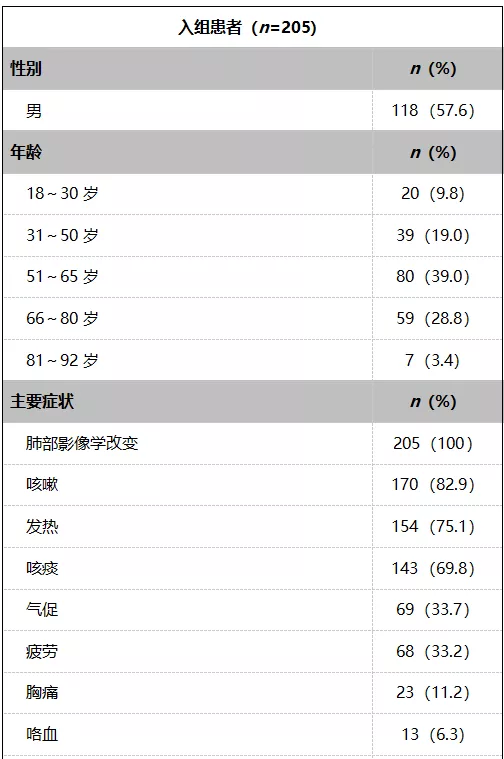
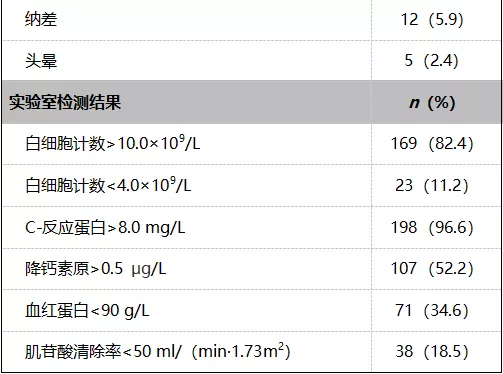
1. 患者入组
研究结果
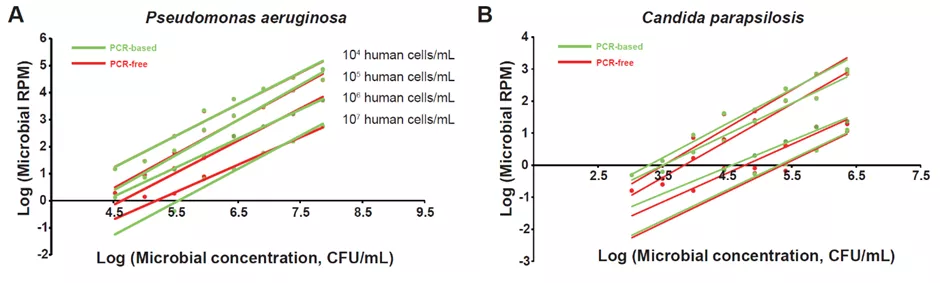
1. 微生物的序列数(RPM)与样本中人细胞浓度成反比
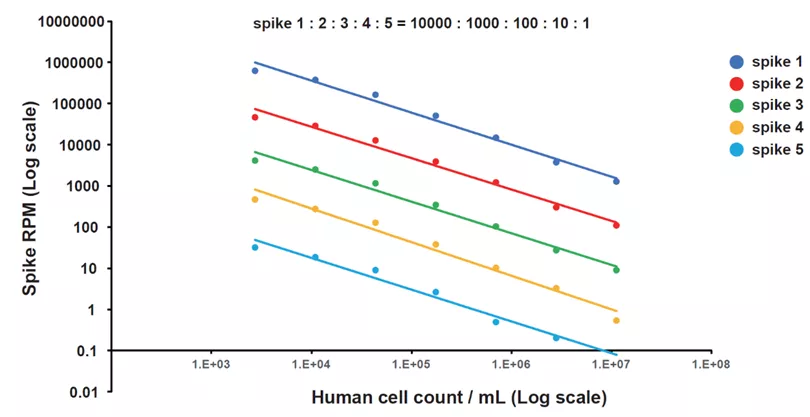
2. Q-mNGS内标分子的筛选与评估
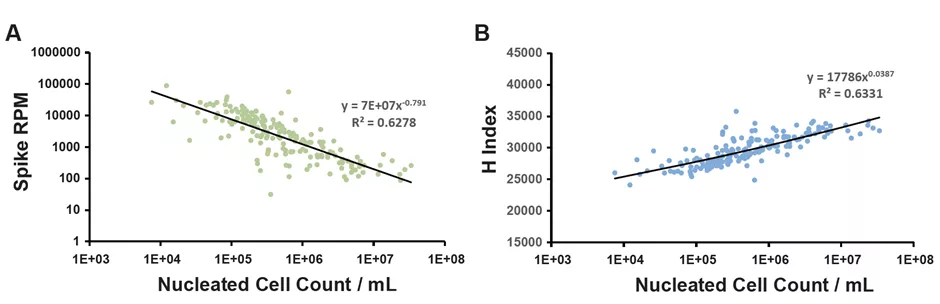
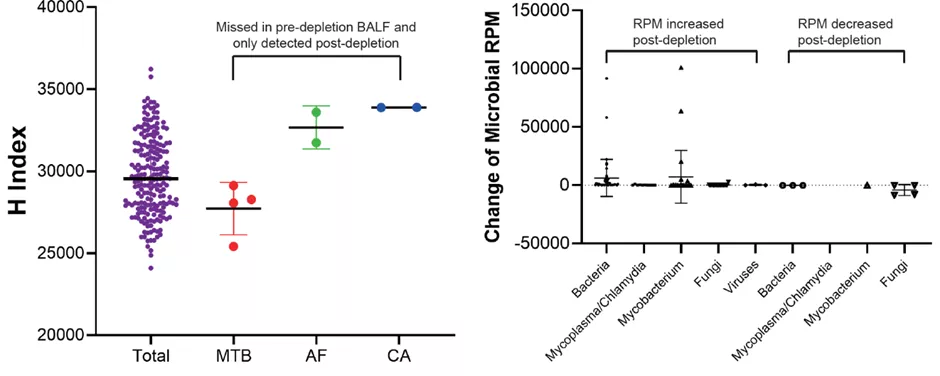
3. 差异化裂解去宿主对mNGS检测性能的影响
图5 差异化裂解去宿主对微生物检测性能的影响
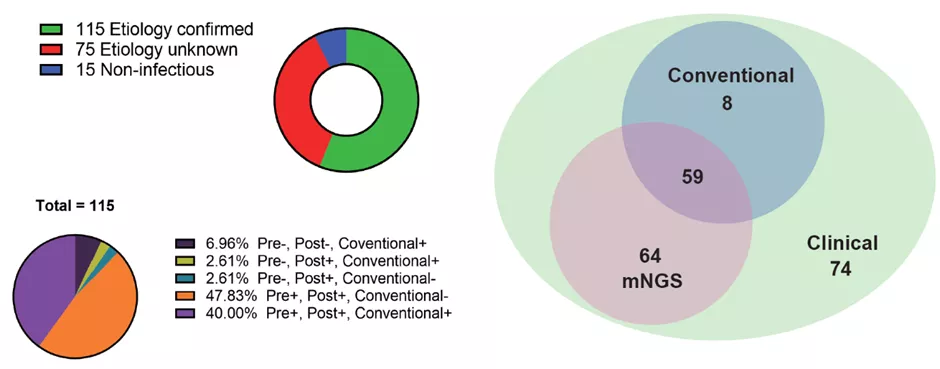
4. Q-mNGS的诊断性能评估
讨论与分析
参考文献
[1]Gu W, Miller S, Chiu CY. Clinical Metagenomic Next-Generation Sequencing for Pathogen Detection[J]. Annu Rev Pathol, 2019, 14:319-338.
[2]李颖,麻锦敏.宏基因组学测序技术在中重症感染中的临床应用专家共识(第一版)[J]. 中华危重病急救医学,2020,32(5):531-536.
[3]Street T L, Barker L, Sanderson N D, et al. Optimizing DNA Extraction Methods for Nanopore Sequencing of Neisseria gonorrhoeae Directly from Urine Samples[J]. J Clin Microbiol, 2020, 58(3):e01822-19.
[4]Charalampous T, Kay GL, Richardson H, et al. Nanopore metagenomics enables rapid clinical diagnosis of bacterial lower respiratory infection[J]. Nat Biotechnol, 2019, 37 7:783-792.
[5]Luan Y, Hu H, Liu C, et al. A proof-of-concept study of an automated solution for clinical metagenomic next-generation sequencing[J]. J Appl Microbiol, 2021, 131(2):1007-1016.
 后可发表评论
后可发表评论

相关推荐
1
肺活检(支气管镜操作)所致大出血的预防与救治
1.2w
2
詹庆元教授:重症肺炎的救治:如何降低病死率?
1.2w
3
李琦教授:重症肺炎早期诊断和危险分层生物标志物研究进展
8999
4
床旁支气管镜在RICU/MICU中的介入治疗
8078
5
一次性支气管镜在RICU有必要吗?
7111
6
俯卧位通气在重症肺炎中的应用进展
6782
7
健康中国 春归武汉|解立新教授专访:M-ROSE在ICU患者肺部真菌感染中的应用
6744
8
重症肺部感染病原学诊断的巨大进步:浅谈二代测序技术
6431
9
黎毅敏教授:重症肺炎的诊治——来自AI的视角
6243
10
呼吸内镜介入技术与呼吸危重症医学发展
6007



友情链接
联系我们
 公众号
公众号
 客服微信
客服微信